Overview
The Thermal Desorption Chemical Ionization Mass Spectrometer (TDCIMS) is an instrument that is capable of measuring the chemical composition of particles as small as 4 nm. It accomplishes this with a sensitivity that makes it possible to measure the molecular composition of nanoparticles at ambient concentrations in the atmosphere. The instrument builds on our experience in developing highly sensitive chemical ionization mass spectrometers for the detection of atmospheric gas-phase compounds at concentrations below 105 molecules/cm3. In our experimental setup, described in detail below, aerosol particles are charged, size-selected, and then collected by electrostatic deposition onto a metal filament. Next, the filament is slid into a chemical ionization region where it is resistively heated to evaporate the sampled particles. The desorbed molecules are ionized at atmospheric pressure by proton transfer with protonated water clusters or oxygen anions. Ions are transferred to a mass spectrometer for mass analysis.
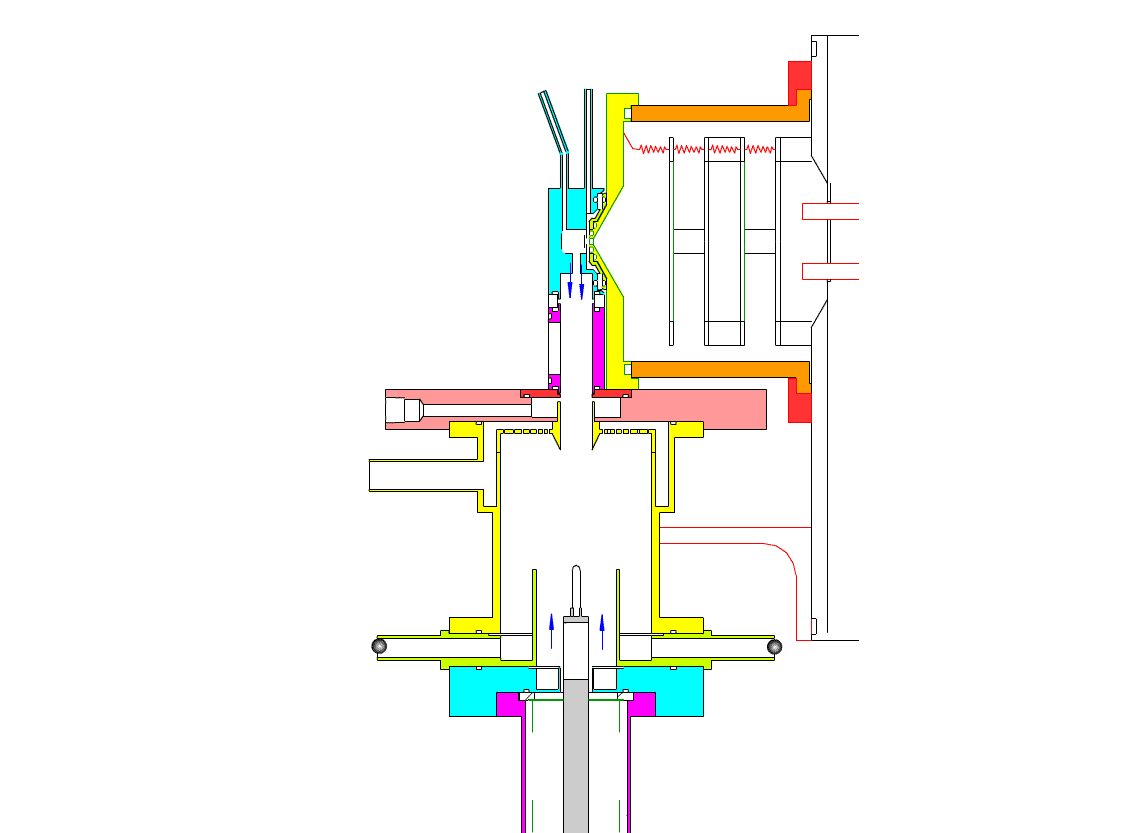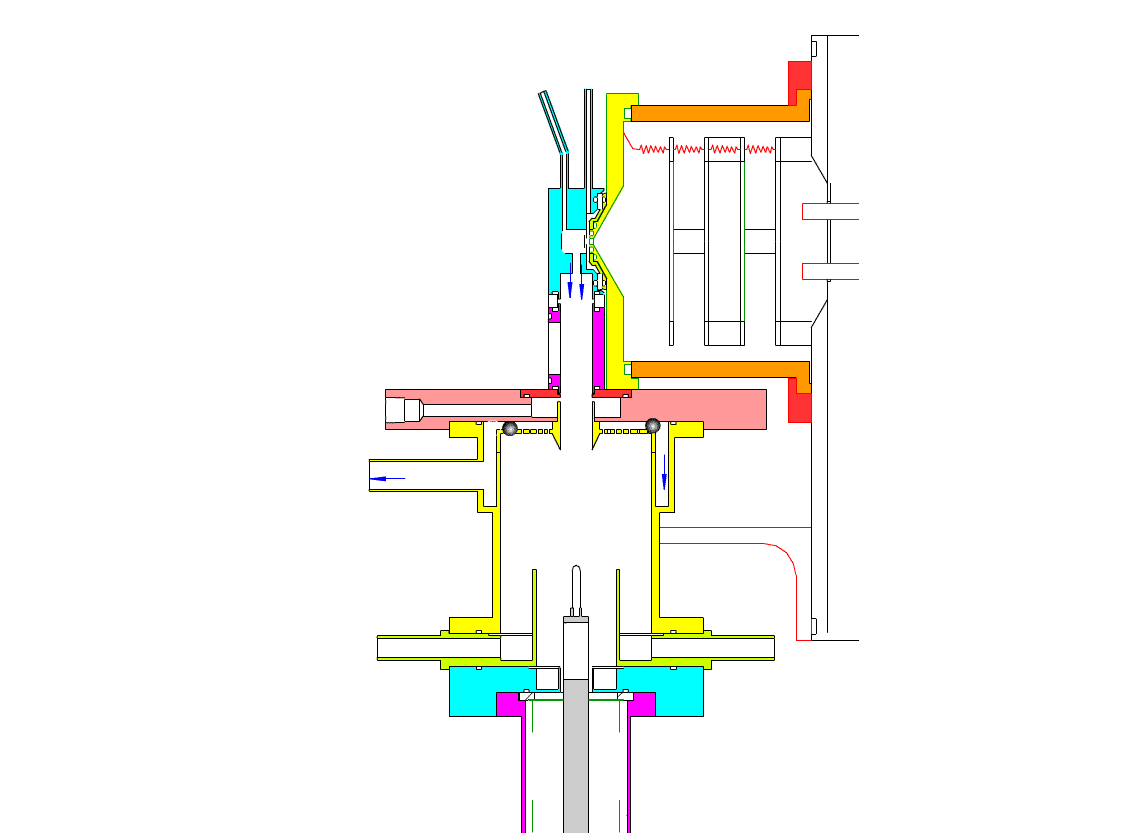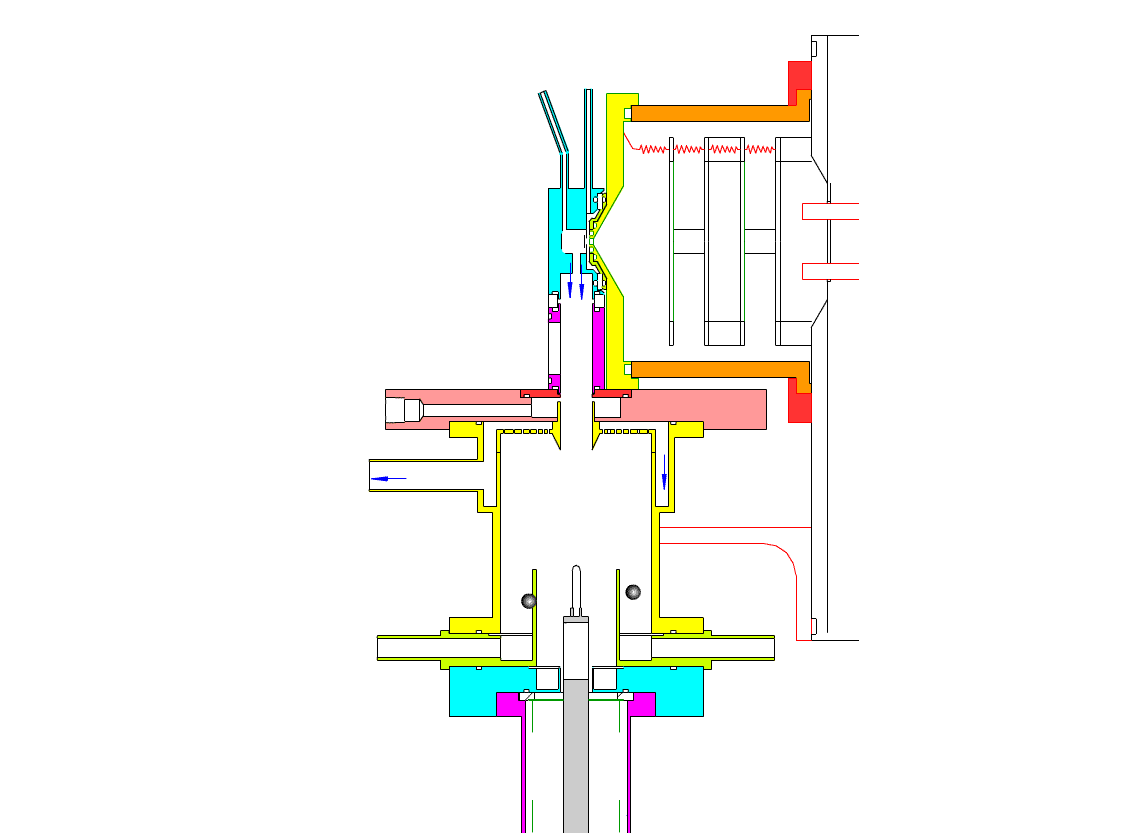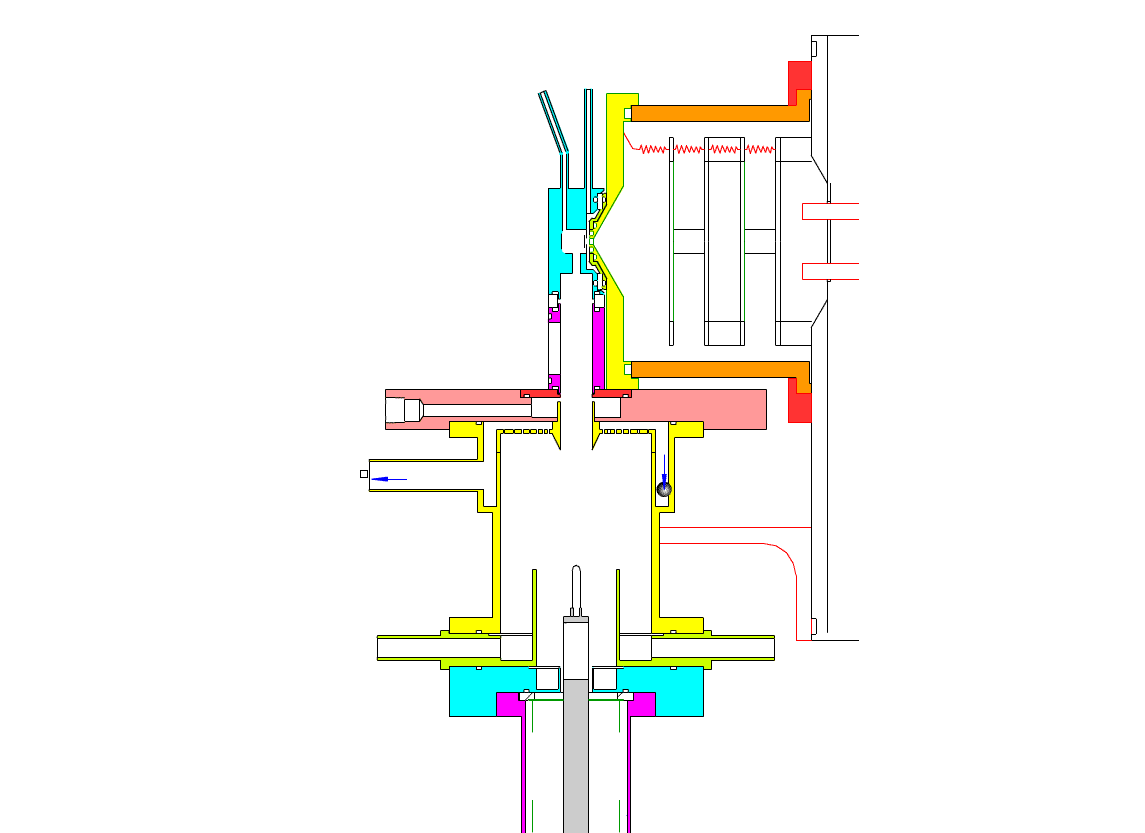
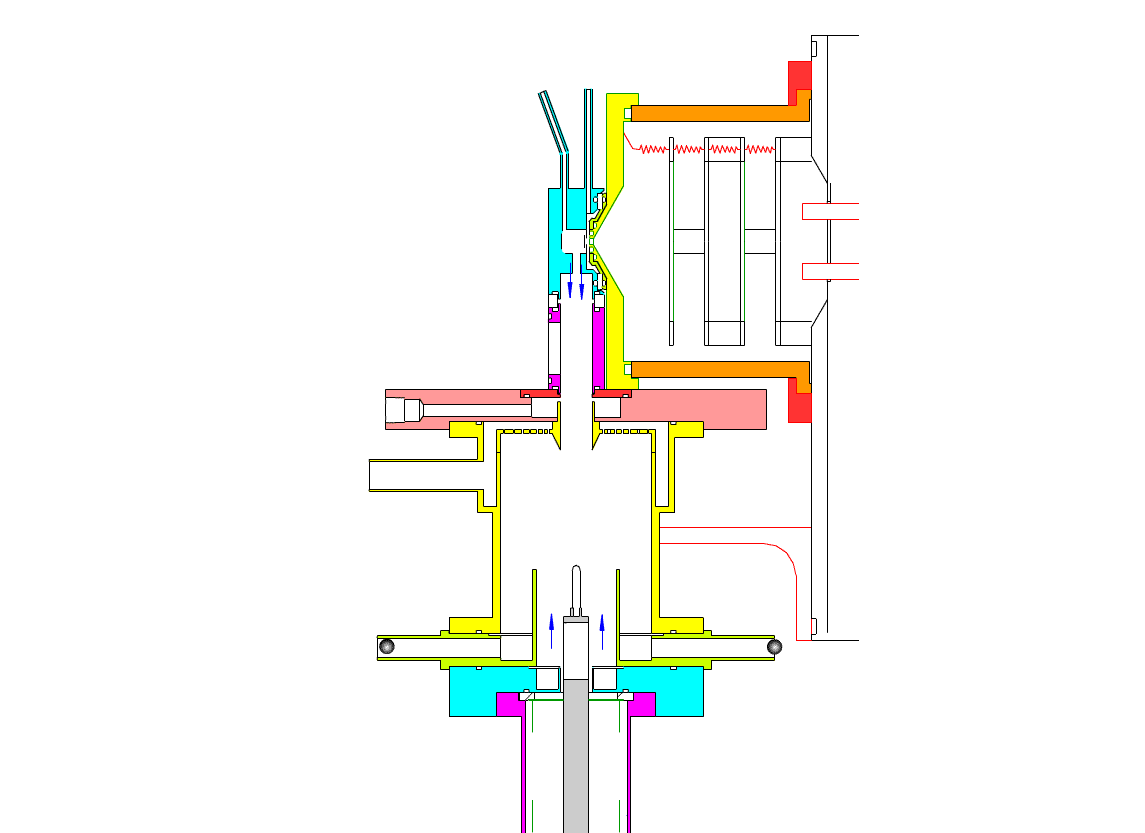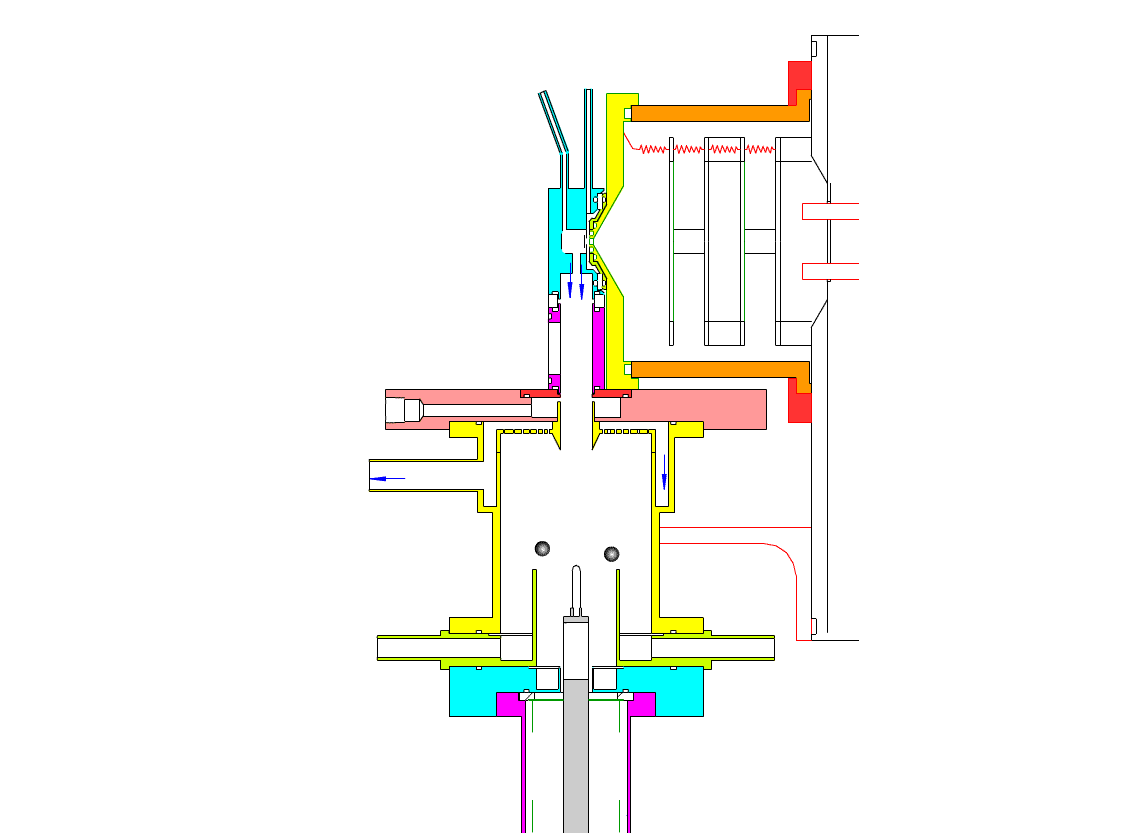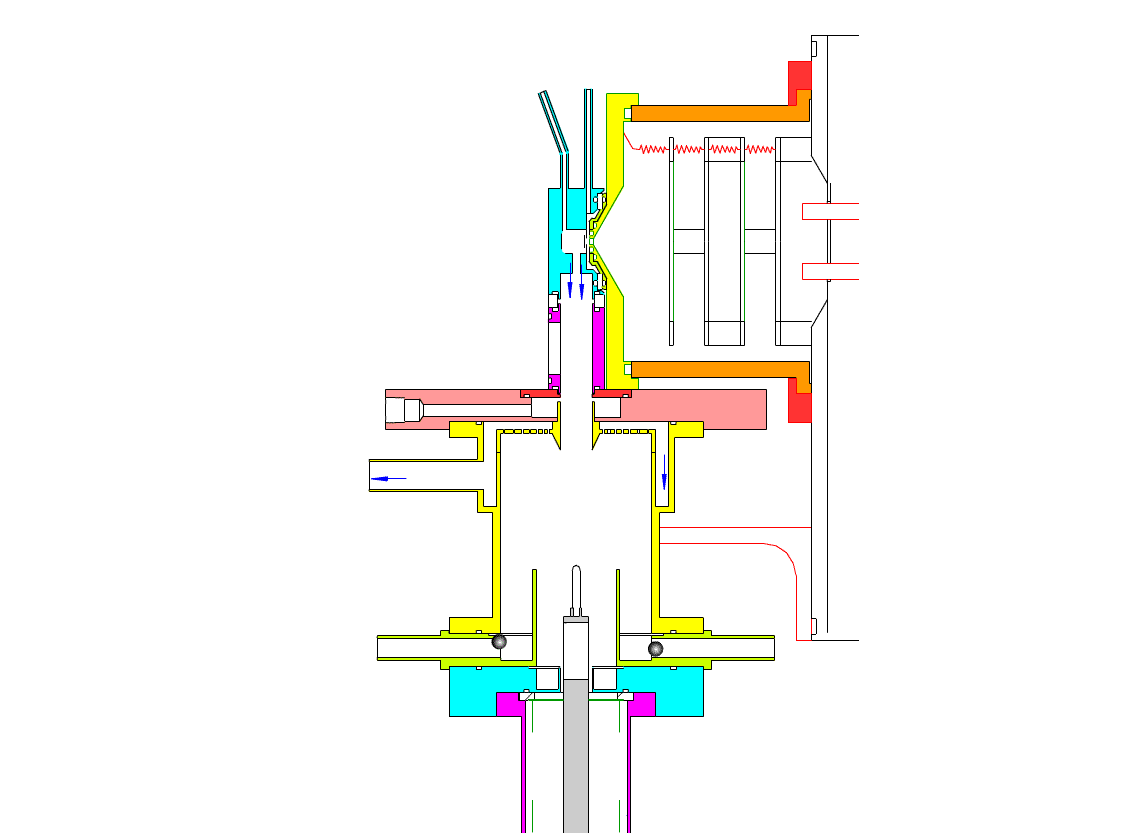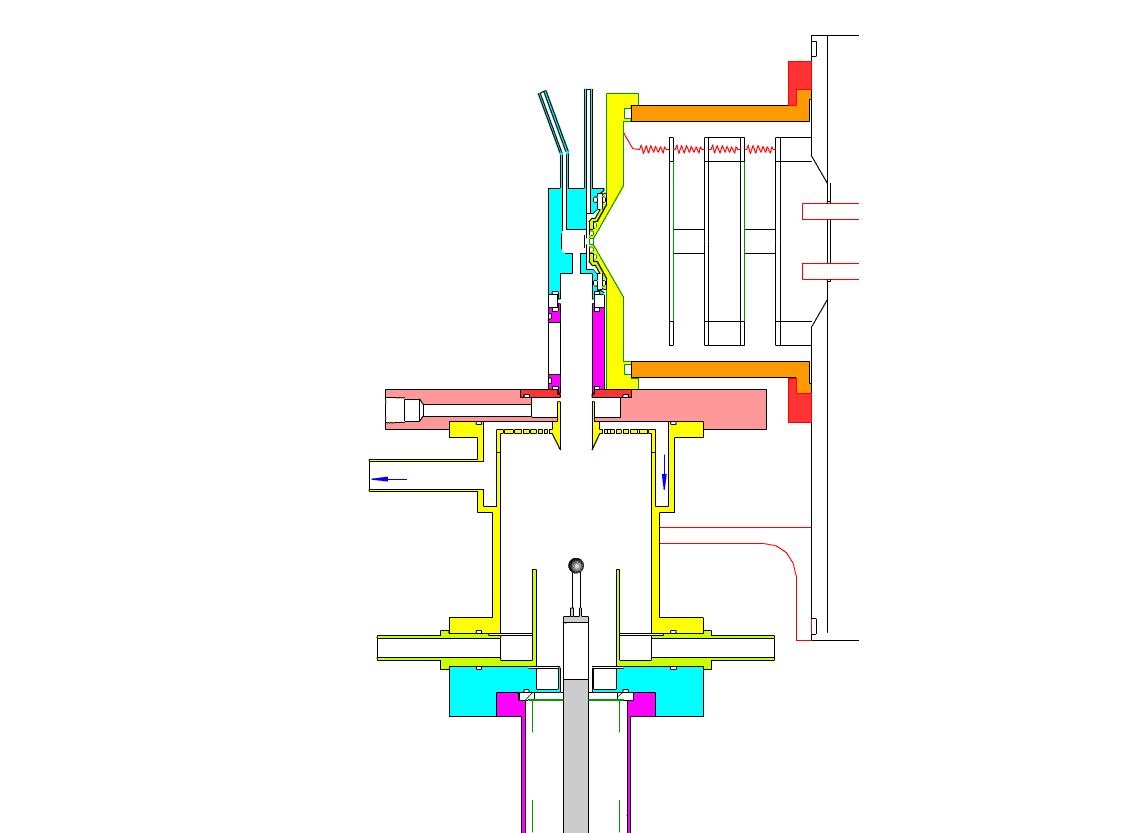

Particle Collection
The electrostatic precipitator consists of a 6 cm inside diameter, 6.5 cm long coaxial collection tube, with a central ceramic rod holding a metal filament made of platinum (Figures 1-3). A flow of air (1-10 sl/m) containing charged ultrafine particles is continuously drawn in through the outer annulus of the collection tube. An inner tube (3 cm diameter) surrounds the collection filament and is flushed with ultra-pure nitrogen, which protects the collection filament from contamination by gaseous compounds present in the particle-laden air flowing in the outer annulus. The combined flow is then drawn out downstream in a manner that insures laminar flow conditions in the collection tube.
In the collection position, the metal filament is either aligned with, or slightly protruding from, the end of the inner sheath tube. The exact position is set within 0.02 cm by a computer controlled linear actuator that also rapidly slides the filament from the collection position into the evaporation-ionization chamber for analysis. Figure 2 shows the precipitator during collection of particles. Here, an electric field is maintained between the outermost stainless steel cylinder and the filament to move charged particles from the sample airflow, through the nitrogen sheath gas and onto the filament. The collection potential on the filament is usually set to ~4000 V, balancing high collection efficiency with the potential for corona emission around the filament at excessive voltages. Particles are collected for a time period that corresponds to the accumulation of a tens of picograms or more on the wire, usually varying 10 – 60 min. After collection, the wire is slid into the evaporation-ionization chamber for analysis (Figure 3). The analysis process is described next.

Sample Desorption and Ionization
This step occurs within the evaporation-ionization chamber, shown above in Figure 4. The evaporation-ionization chamber is a small half-cylindrical region of about 1.5 cm3 in volume. The wall of the chamber is lined with a Am241 radioactive foil, which emits alpha particles that ionize the buffer gas mixture to form nitrogen and oxygen ions that subsequently react to form H3O+, O2–, and their clusters with water, as the primary stable reagent ions. These ions will then react with the compounds evaporated from the aerosol. We can use these two ion chemistries to detect the following compounds in aerosol particles:
- cationic nitrogen bases (ammonia, amines)
- inorganic acids (nitric acid, sulfuric acid)
- organic acids
- oxygenated and unsaturated organics
The ion source assembly is temperature controlled to 50 °C to avoid condensation of the evaporated material back onto the walls. A nitrogen flow is fed into the evaporation-ionization chamber and serves two purposes. The first is to deliver the reagent ions and flush the ionization chamber, pushing evaporated compounds slowly towards the vacuum system aperture. The second purpose of this flow is to isolate the ion source from the electrostatic precipitator and the sample air, thus minimizing contamination of the ion source. The collection filament can be resistively heated by applying a programmed AC (alternating current) through it. Filament temperature vs. applied current has been measured in a separate test by spot welding a small thermocouple to the filament. When current is being applied to the filament during sample desorption. the filament and chamber are electrically biased to force ions to travel to the vacuum entrance aperture. These and all mass spectrometer lenses are computer controlled, allowing automated switching from positive to negative ion measurement.
Calibration

Figure 5. TDCIMS ion signals during analysis of ammonium sulfate nanoparticles. The two panels to the left show sample desorption peaks, whereas the two panels to the right show background signals.Figure 5, above, shows typical ion peaks for ammonium sulfate calibration aerosol. The collected aerosol mass for the ammonium peak is 3.2 pg, corresponding to 14 nm particles collected at a concentration of 2750 cm-3 for 60 s. Tests were also performed in negative ion mode to establish instrument response for sulfate in ammonium sulfate aerosols. A peak is observed for bisulfate (the stable form of sulfate in the gas phase) when the filament is heated in the ionization chamber (lower two plots in Figure 5). The data for bisulfate corresponds to 10 nm particles being collected at a concentration of 4300 cm-3 for 120 s, resulting a collected aerosol mass of 1.8 pg. In the case of bisulfate ion, the bottom figure shows two peaks arising from sample desorption and ionization. The presence of two peaks suggests two separate mechanisms for the formation of bisulfate from ammonium sulfate aerosol. The signal to noise ratio for the bisulfate trace in the bottom figure is 60.

A series of tests were made to compare ion peak integrated area to the collected aerosol mass. For these experiments, ammonium sulfate aerosols were generated and size-classified into 14 and 10 nm diameter aerosol particles for measurements of ammonium and sulfate, respectively. Collection times were varied from 15 s to 5 min to vary the collected aerosol mass. Plots of integrated ion peak area versus collected aerosol mass are shown in Figure 6. In the plot, uncertainties associated with generating ammonium sulfate particles of known diameter were primarily responsible for the error in the total aerosol mass. The uncertainty in the measurement of the integrated peak area arises from the manual integration scheme that we employed for this study. The integrated peak areas shown in Figure 6 are the net areas obtained by subtracting the background peak areas from that of the collected aerosols. The data thus represent the chemical compounds present only in the particulate phase. For both ammonium and sulfate, the trends are linear. In the case of bisulfate (bottom plot), a slight departure from linearity can be seen for collected aerosol mass below 1 pg. Although the cause of this departure from linearity is not known, since it occurs near the detection limit we can state a effective detection limit (1 pg) that corresponds to the lowest mass for which fully linear calibration is observed.