Welcome to the Marazzi Lab!
The Marazzi Laboratory studies epigenetic and chromatin-mediated control of gene expression. We are interested in how mutations cause infection, inflammatory and neurodegenerative diseases. We use biochemistry, genetics, and next generation sequencing techniques to understand molecular mechanisms and genome-wide effects of known and novel candidate genes that cause juvenile forms of disease. We care about disease of young age. For reference, please see our recent works on neurodegeneration, inflammation and infection.
Papers
Nuclear RNA catabolism controls endogenous retroviruses, gene expression asymmetry, and dedifferentiation

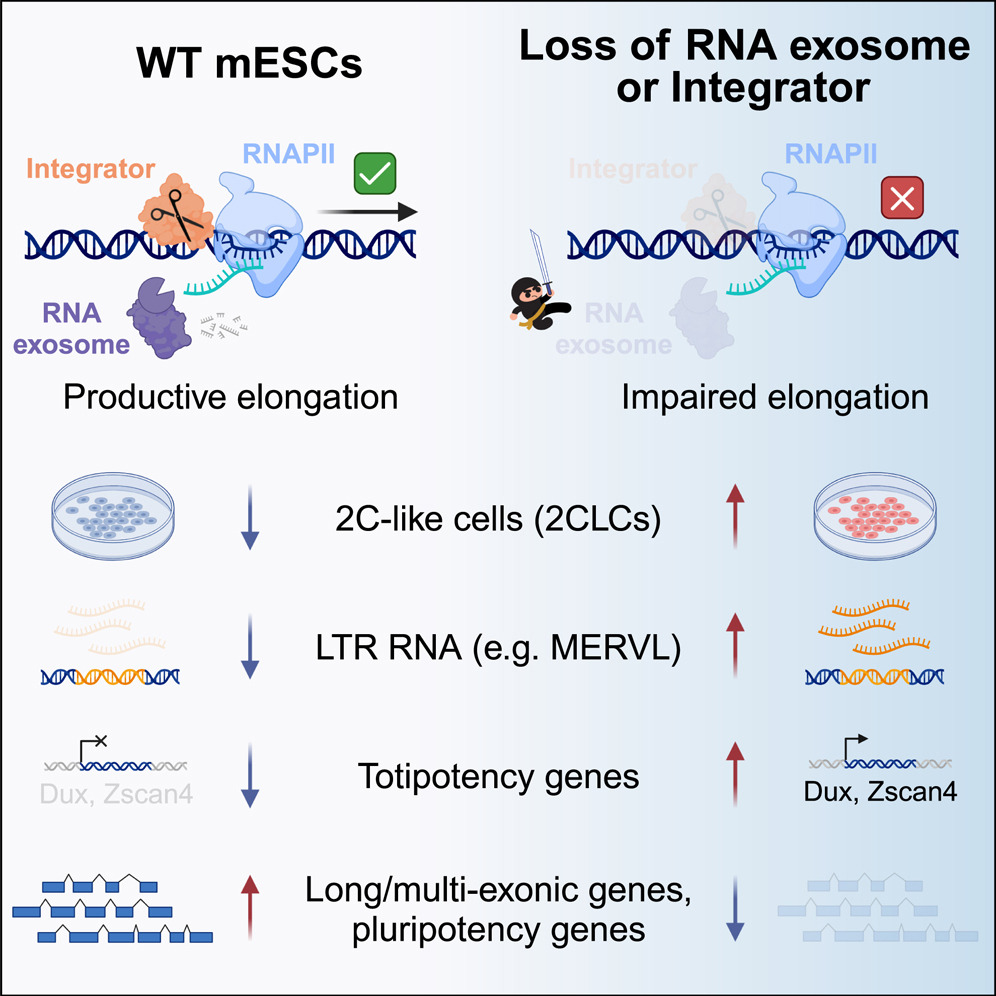
- The nRE and INT maintain ES identity
- Loss of nRE and INT induce a 2C-like totipotent cell state
- nRE and INT suppress ERV expression
- nRE and INT suppress non-productive transcription
Endogenous retroviruses (ERVs) are remnants of ancient parasitic infections and comprise sizable portions of most genomes. Although epigenetic mechanisms silence most ERVs by generating a repressive environment that prevents their expression (heterochromatin), little is known about mechanisms silencing ERVs residing in open regions of the genome (euchromatin). This is particularly important during embryonic development, where induction and repression of distinct classes of ERVs occur in short temporal windows. Here, we demonstrate that transcription-associated RNA degradation by the nuclear RNA exosome and Integrator is a regulatory mechanism that controls the productive transcription of most genes and many ERVs involved in preimplantation development. Disrupting nuclear RNA catabolism promotes dedifferentiation to a totipotent-like state characterized by defects in RNAPII elongation and decreased expression of long genes (gene-length asymmetry). Our results indicate that RNA catabolism is a core regulatory module of gene networks that safeguards RNAPII activity, ERV expression, cell identity, and developmental potency.
Generation of host-directed and virus-specific antivirals using targeted protein degradation promoted by small molecules and viral RNA mimics

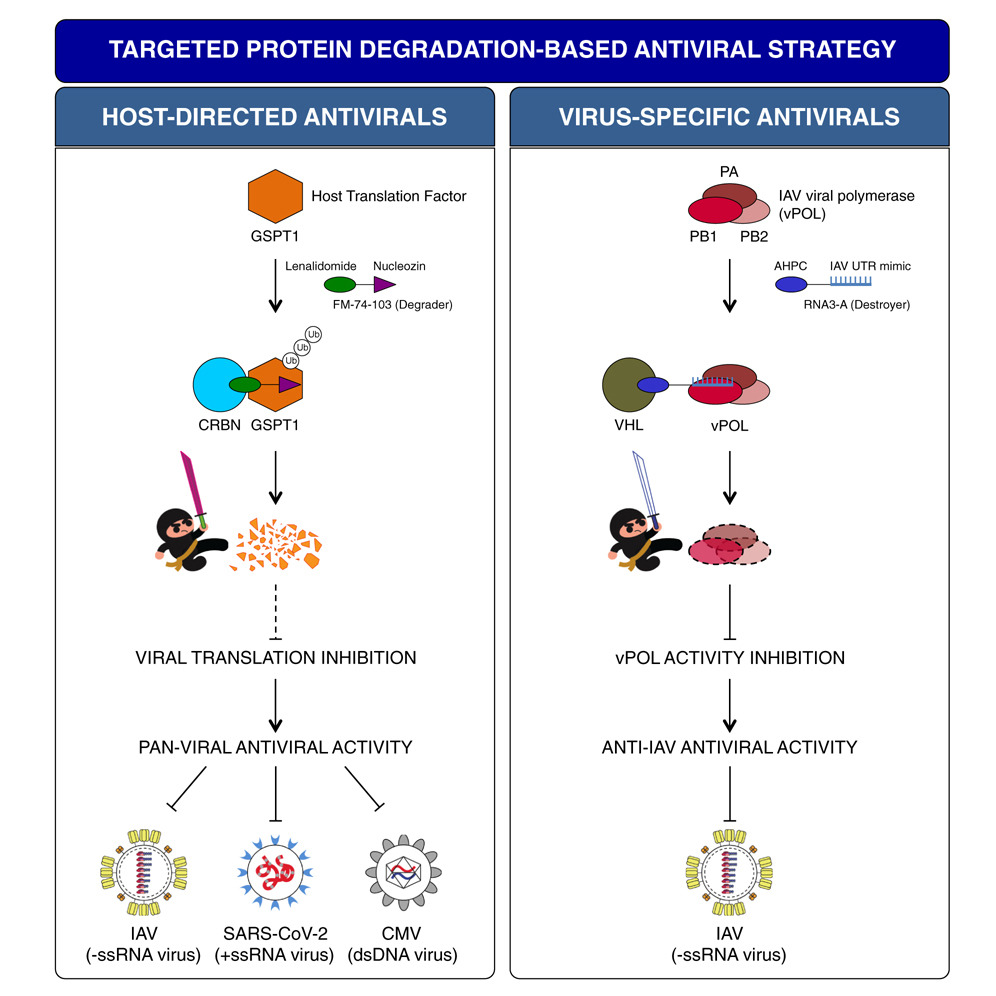
- FM-74-103 inhibits IAV, SARS-CoV-2, and CMV infections through GSPT1 degradation
- GSPT1 degradation is antiviral as it is a crucial host factor for many viruses
- Destroyers are viral RNA mimics that target viral proteins for degradation
- Targeting viral polymerase with Destroyers is a virus-specific antiviral strategy
Targeted protein degradation (TPD), as exemplified by proteolysis-targeting chimera (PROTAC), is an emerging drug discovery platform. PROTAC molecules, which typically contain a target protein ligand linked to an E3 ligase ligand, recruit a target protein to the E3 ligase to induce its ubiquitination and degradation. Here, we applied PROTAC approaches to develop broad-spectrum antivirals targeting key host factors for many viruses and virus-specific antivirals targeting unique viral proteins. For host-directed antivirals, we identified a small-molecule degrader, FM-74-103, that elicits selective degradation of human GSPT1, a translation termination factor. FM-74-103-mediated GSPT1 degradation inhibits both RNA and DNA viruses. Among virus-specific antivirals, we developed viral RNA oligonucleotide-based bifunctional molecules (Destroyers). As a proof of principle, RNA mimics of viral promoter sequences were used as heterobifunctional molecules to recruit and target influenza viral polymerase for degradation. This work highlights the broad utility of TPD to rationally design and develop next-generation antivirals.
Clonally expanded CD8 T cells characterize amyotrophic lateral sclerosis-4
 Amyotrophic lateral sclerosis (ALS) is a heterogenous neurodegenerative disorder that affects motor neurons and voluntary muscle control1. ALS heterogeneity includes the age of manifestation, the rate of progression and the anatomical sites of symptom onset. Disease-causing mutations in specific genes have been identified and define different subtypes of ALS1. Although several ALS-associated genes have been shown to affect immune functions2, whether specific immune features account for ALS heterogeneity is poorly understood. Amyotrophic lateral sclerosis-4 (ALS4) is characterized by juvenile onset and slow progression3. Patients with ALS4 show motor difficulties by the time that they are in their thirties, and most of them require devices to assist with walking by their fifties. ALS4 is caused by mutations in the senataxin gene (SETX). Here, using Setx knock-in mice that carry the ALS4-causative L389S mutation, we describe an immunological signature that consists of clonally expanded, terminally differentiated effector memory (TEMRA) CD8 T cells in the central nervous system and the blood of knock-in mice. Increased frequencies of antigen-specific CD8 T cells in knock-in mice mirror the progression of motor neuron disease and correlate with anti-glioma immunity. Furthermore, bone marrow transplantation experiments indicate that the immune system has a key role in ALS4 neurodegeneration. In patients with ALS4, clonally expanded TEMRA CD8 T cells circulate in the peripheral blood. Our results provide evidence of an antigen-specific CD8 T cell response in ALS4, which could be used to unravel disease mechanisms and as a potential biomarker of disease state.
Amyotrophic lateral sclerosis (ALS) is a heterogenous neurodegenerative disorder that affects motor neurons and voluntary muscle control1. ALS heterogeneity includes the age of manifestation, the rate of progression and the anatomical sites of symptom onset. Disease-causing mutations in specific genes have been identified and define different subtypes of ALS1. Although several ALS-associated genes have been shown to affect immune functions2, whether specific immune features account for ALS heterogeneity is poorly understood. Amyotrophic lateral sclerosis-4 (ALS4) is characterized by juvenile onset and slow progression3. Patients with ALS4 show motor difficulties by the time that they are in their thirties, and most of them require devices to assist with walking by their fifties. ALS4 is caused by mutations in the senataxin gene (SETX). Here, using Setx knock-in mice that carry the ALS4-causative L389S mutation, we describe an immunological signature that consists of clonally expanded, terminally differentiated effector memory (TEMRA) CD8 T cells in the central nervous system and the blood of knock-in mice. Increased frequencies of antigen-specific CD8 T cells in knock-in mice mirror the progression of motor neuron disease and correlate with anti-glioma immunity. Furthermore, bone marrow transplantation experiments indicate that the immune system has a key role in ALS4 neurodegeneration. In patients with ALS4, clonally expanded TEMRA CD8 T cells circulate in the peripheral blood. Our results provide evidence of an antigen-specific CD8 T cell response in ALS4, which could be used to unravel disease mechanisms and as a potential biomarker of disease state.
TOP1 inhibition therapy protects against SARS-CoV-2-induced lethal inflammation

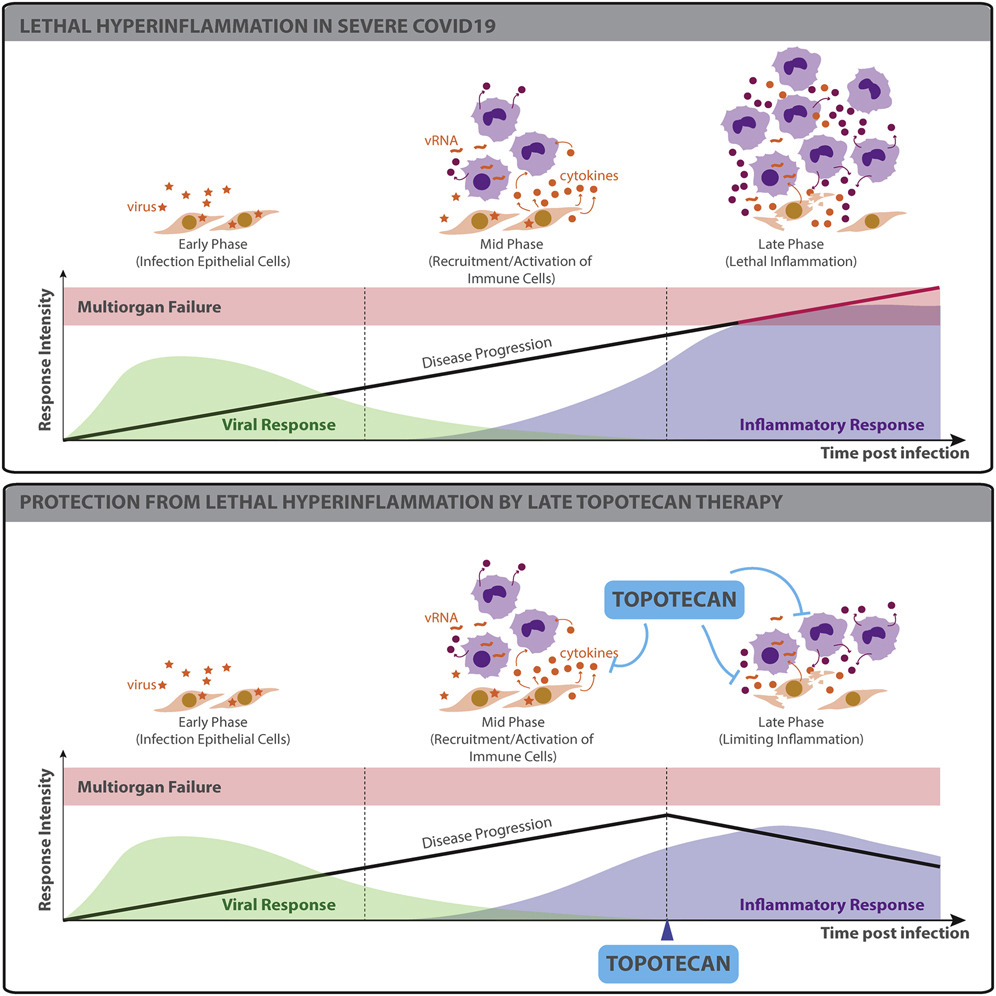
- TOP1 activates inflammatory gene expression during SARS-CoV-2 infection
- Topotecan (TPT), a TOP1 FDA-approved drug, suppresses inflammatory gene expression
- Inflammatory genes suppressed by TPT are overexpressed in COVID-19 lung autopsies
- TPT therapy protects from hyper-inflammation and death in preclinical models
The ongoing pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is currently affecting millions of lives worldwide. Large retrospective studies indicate that an elevated level of inflammatory cytokines and pro-inflammatory factors are associated with both increased disease severity and mortality. Here, using multidimensional epigenetic, transcriptional, in vitro, and in vivo analyses, we report that topoisomerase 1 (TOP1) inhibition suppresses lethal inflammation induced by SARS-CoV-2. Therapeutic treatment with two doses of topotecan (TPT), an FDA-approved TOP1 inhibitor, suppresses infection-induced inflammation in hamsters. TPT treatment as late as 4 days post-infection reduces morbidity and rescues mortality in a transgenic mouse model. These results support the potential of TOP1 inhibition as an effective host-directed therapy against severe SARS-CoV-2 infection. TPT and its derivatives are inexpensive clinical-grade inhibitors available in most countries. Clinical trials are needed to evaluate the efficacy of repurposing TOP1 inhibitors for severe coronavirus disease 2019 (COVID-19) in humans.
Hybrid Gene Origination Creates Human-Virus Chimeric Proteins during Infection

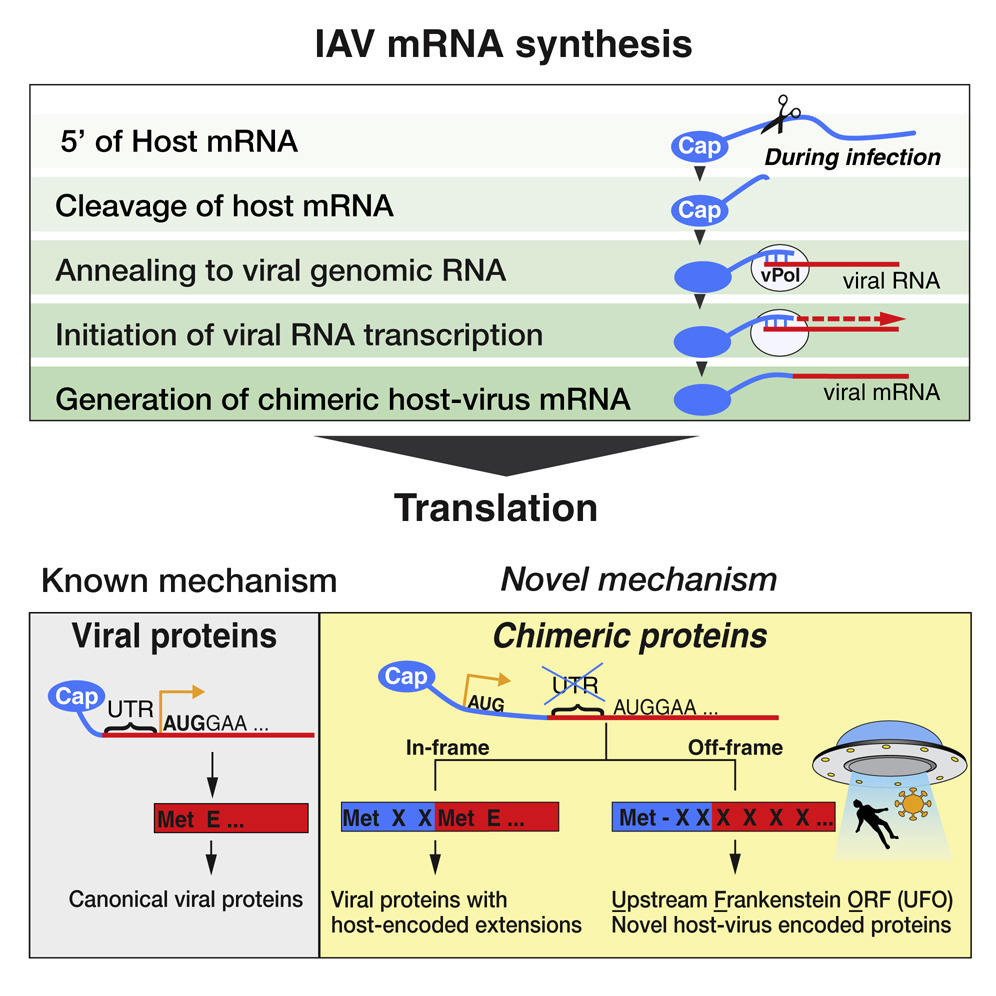
- A mechanism of hybrid gene birth is employed by many families of RNA viruses
- Human RNA and viral RNA encode new genes together
- Hybrid genes either make extensions of viral proteins or novel proteins (UFOs)
- Human-virus genes and proteins play roles in pathogenesis and are conserved
RNA viruses are a major human health threat. The life cycles of many highly pathogenic RNA viruses like influenza A virus (IAV) and Lassa virus depends on host mRNA, because viral polymerases cleave 5′-m7G-capped host transcripts to prime viral mRNA synthesis (“cap-snatching”). We hypothesized that start codons within cap-snatched host transcripts could generate chimeric human-viral mRNAs with coding potential. We report the existence of this mechanism of gene origination, which we named “start-snatching.” Depending on the reading frame, start-snatching allows the translation of host and viral “untranslated regions” (UTRs) to create N-terminally extended viral proteins or entirely novel polypeptides by genetic overprinting. We show that both types of chimeric proteins are made in IAV-infected cells, generate T cell responses, and contribute to virulence. Our results indicate that during infection with IAV, and likely a multitude of other human, animal and plant viruses, a host-dependent mechanism allows the genesis of hybrid genes.
The RNA Exosome Syncs IAV-RNAPII Transcription to Promote Viral Ribogenesis and Infectivity


- The nuclear RNA exosome coordinates viral polymerase and RNAPII transcription
- The nuclear RNA exosome enhances host-viral RNA hybrids, chimeras, and viral ribogenesis
- RNA exosome loss-of-function in epithelial and immune cells impairs viral growth
- Broken symmetry hypothesis: Viruses target genes whose mutations lead to disease
The nuclear RNA exosome is an essential multi-subunit complex that controls RNA homeostasis. Congenital mutations in RNA exosome genes are associated with neurodegenerative diseases. Little is known about the role of the RNA exosome in the cellular response to pathogens. Here, using NGS and human and mouse genetics, we show that influenza A virus (IAV) ribogenesis and growth are suppressed by impaired RNA exosome activity. Mechanistically, the nuclear RNA exosome coordinates the initial steps of viral transcription with RNAPII at host promoters. The viral polymerase complex co-opts the nuclear RNA exosome complex and cellular RNAs en route to 3′ end degradation. Exosome deficiency uncouples chromatin targeting of the viral polymerase complex and the formation of cellular:viral RNA hybrids, which are essential RNA intermediates that license transcription of antisense genomic viral RNAs. Our results suggest that evolutionary arms races have shaped the cellular RNA quality control machinery.
Targeting Viral Proteostasis Limits Influenza Virus, HIV, and Dengue Virus Infection

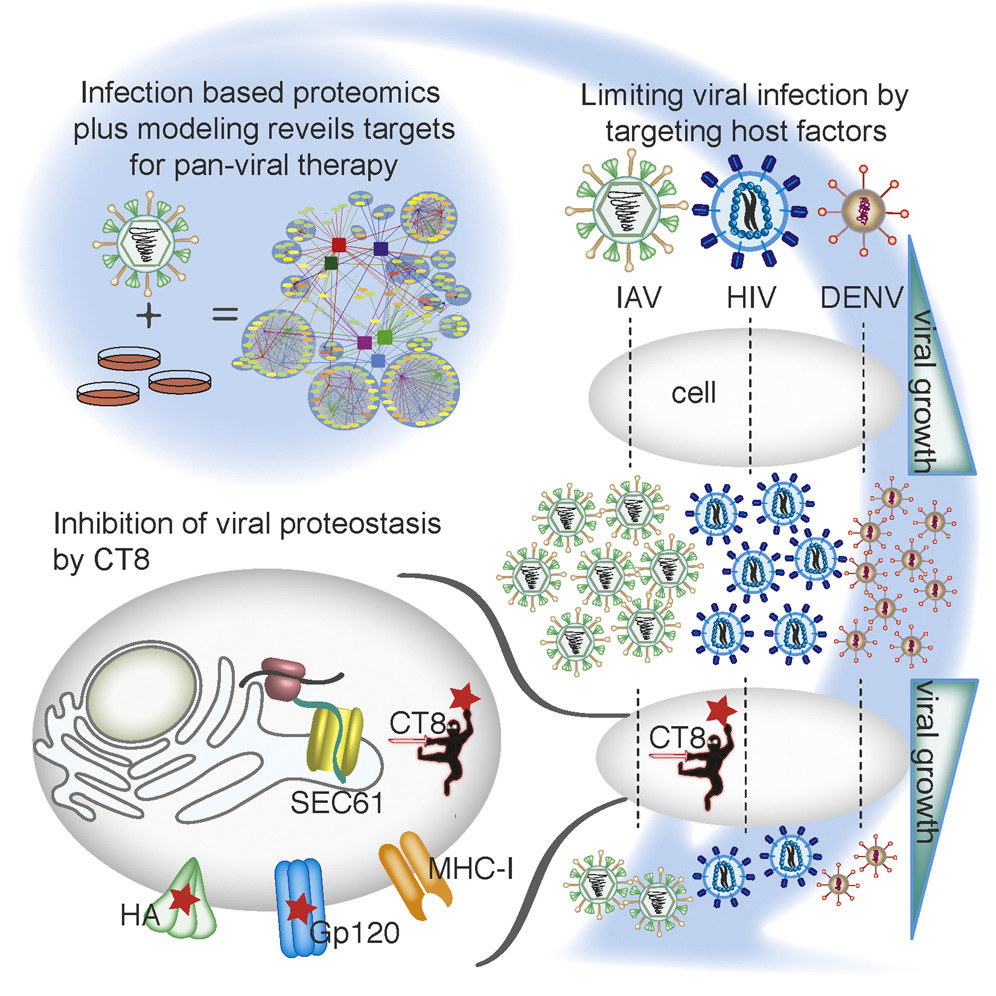
- Generated replication-competent, tagged influenza viruses
- Constructed human-influenza interactome network during an infection
- Mathematical modeling revealed host targets for pan-viral inhibition
- Sec61 inhibition alters viral proteostasis and suppresses viral replication
Viruses are obligate parasites and thus require the machinery of the host cell to replicate. Inhibition of host factors co-opted during active infection is a strategy hosts use to suppress viral replication and a potential pan-antiviral therapy. To define the cellular proteins and processes required for a virus during infection is thus crucial to understanding the mechanisms of virally induced disease. In this report, we generated fully infectious tagged influenza viruses and used infection-based proteomics to identify pivotal arms of cellular signaling required for influenza virus growth and infectivity. Using mathematical modeling and genetic and pharmacologic approaches, we revealed that modulation of Sec61-mediated cotranslational translocation selectively impaired glycoprotein proteostasis of influenza as well as HIV and dengue viruses and led to inhibition of viral growth and infectivity. Thus, by studying virus-human protein-protein interactions in the context of active replication, we have identified targetable host factors for broad-spectrum antiviral therapies.
Senataxin suppresses the antiviral transcriptional response and controls viral biogenesis
 The human helicase senataxin (SETX) has been linked to the neurodegenerative diseases amyotrophic lateral sclerosis (ALS4) and ataxia with oculomotor apraxia (AOA2). Here we identified a role for SETX in controlling the antiviral response. Cells that had undergone depletion of SETX and SETX-deficient cells derived from patients with AOA2 had higher expression of antiviral mediators in response to infection than did wild-type cells. Mechanistically, we propose a model whereby SETX attenuates the activity of RNA polymerase II (RNAPII) at genes stimulated after a virus is sensed and thus controls the magnitude of the host response to pathogens and the biogenesis of various RNA viruses (e.g., influenza A virus and West Nile virus). Our data indicate a potentially causal link among inborn errors in SETX, susceptibility to infection and the development of neurologic disorders.
The human helicase senataxin (SETX) has been linked to the neurodegenerative diseases amyotrophic lateral sclerosis (ALS4) and ataxia with oculomotor apraxia (AOA2). Here we identified a role for SETX in controlling the antiviral response. Cells that had undergone depletion of SETX and SETX-deficient cells derived from patients with AOA2 had higher expression of antiviral mediators in response to infection than did wild-type cells. Mechanistically, we propose a model whereby SETX attenuates the activity of RNA polymerase II (RNAPII) at genes stimulated after a virus is sensed and thus controls the magnitude of the host response to pathogens and the biogenesis of various RNA viruses (e.g., influenza A virus and West Nile virus). Our data indicate a potentially causal link among inborn errors in SETX, susceptibility to infection and the development of neurologic disorders.
Review Articles
Unconventional viral gene expression mechanisms as therapeutic targets
 Unlike the human genome that comprises mostly noncoding and regulatory sequences, viruses have evolved under the constraints of maintaining a small genome size while expanding the efficiency of their coding and regulatory sequences. As a result, viruses use strategies of transcription and translation in which one or more of the steps in the conventional gene–protein production line are altered. These alternative strategies of viral gene expression (also known as gene recoding) can be uniquely brought about by dedicated viral enzymes or by co-opting host factors (known as host dependencies). Targeting these unique enzymatic activities and host factors exposes vulnerabilities of a virus and provides a paradigm for the design of novel antiviral therapies. In this Review, we describe the types and mechanisms of unconventional gene and protein expression in viruses, and provide a perspective on how future basic mechanistic work could inform translational efforts that are aimed at viral eradication.
Unlike the human genome that comprises mostly noncoding and regulatory sequences, viruses have evolved under the constraints of maintaining a small genome size while expanding the efficiency of their coding and regulatory sequences. As a result, viruses use strategies of transcription and translation in which one or more of the steps in the conventional gene–protein production line are altered. These alternative strategies of viral gene expression (also known as gene recoding) can be uniquely brought about by dedicated viral enzymes or by co-opting host factors (known as host dependencies). Targeting these unique enzymatic activities and host factors exposes vulnerabilities of a virus and provides a paradigm for the design of novel antiviral therapies. In this Review, we describe the types and mechanisms of unconventional gene and protein expression in viruses, and provide a perspective on how future basic mechanistic work could inform translational efforts that are aimed at viral eradication.
Interactions of NF-kappaB with chromatin: the art of being at the right place at the right time
 Transcription factors of the NF-kappaB family are essential regulators of the inflammatory and immune responses. The main ‘switch’ in NF-kappaB activation is cytoplasmic and leads to the release of NF-kappaB proteins from IkappaB molecules, specific inhibitors that prevent their nuclear accumulation. However, it is becoming increasingly apparent that in addition to this required activation step, both recruitment of NF-kappaB to target genes and NF-kappaB-induced transcriptional events after recruitment are actively controlled. Regulated recruitment of NF-kappaB to chromatin generates kinetic complexity in NF-kappaB-dependent gene induction and ‘wires’ NF-kappaB-regulated gene activity to simultaneously activated pathways and transcription factors.
Transcription factors of the NF-kappaB family are essential regulators of the inflammatory and immune responses. The main ‘switch’ in NF-kappaB activation is cytoplasmic and leads to the release of NF-kappaB proteins from IkappaB molecules, specific inhibitors that prevent their nuclear accumulation. However, it is becoming increasingly apparent that in addition to this required activation step, both recruitment of NF-kappaB to target genes and NF-kappaB-induced transcriptional events after recruitment are actively controlled. Regulated recruitment of NF-kappaB to chromatin generates kinetic complexity in NF-kappaB-dependent gene induction and ‘wires’ NF-kappaB-regulated gene activity to simultaneously activated pathways and transcription factors.